数学を取り入れたシミュレーションで材料設計を加速-グラフェン触媒設計に従来法よりも10億倍速い計算時間で成功-
発表のポイント
- カーボンネットワークを単純化した数学モデルの構築に成功。
- 数学を用いた構造の数値化により貴金属フリーのグラフェン触媒の設計が可能に。
- もっとも正確とされる理論計算手法と定性的によく一致したシミュレーション結果が10億倍速く得られ、先端材料候補の探索が加速されることに期待。
概要
3次元の立体的構造を持つカーボンネットワークは、平面構造を持つ2次元グラフェンシート(*1)を曲げることで形成され、シートをうまく曲げると2次元グラフェンとは異なる優れた特性を持つことが知られています。しかし、3次元のカーボンネットワークがなぜ優れた特性を示すのかは系統的に理解されておらず、局所構造と材料全体の特性を結び付けた網羅的な理解が必要とされています。
東北大学材料科学高等研究所の小谷元子教授らの研究グループは、カーボンネットワークをバネで連結された数理モデルとみなし、構造を単純化することで、従来法よりも10億倍速いシミュレーション計算時間で貴金属を使用しない優れた触媒の設計法を見出しました。数学によって提案されたカーボンネットワークを実験的に作製し、その触媒能力を検証することで、数学を用いた材料設計の有用性を実証しました。
本研究成果により、局所構造が持つ幾何学構造の歪みや不安定性などの、これまで難しかった数値化が可能となり、それらが全体の特性に与える影響の理解が可能となりました。この数学手法と従来手法を相補的に組み合わせた大規模シミュレーションと様々な用途への展開が可能な材料開発が進むことが期待されます。
本研究は国際科学雑誌「Carbon」に2021年6月4日(現地時間)付けでオンライン掲載されました。
研究の背景
近年、立体的な3次元構造を持つカーボンネットワーク材料(以下、3次元グラフェンと呼ぶ)が多種多様な特性を示したことから、応用研究が盛んに展開されています。2次元のグラフェンシートを曲げて3次元構造を形成した3次元グラフェンは、2次元グラフェンより優れた特性を示すことが知られています。さらに、それに窒素などの化学元素をドープ(*2)すると、入手しやすい安価な炭素材料で貴金属を使用しない水素製造用触媒になるなど役に立ちかつ優れた機能を持つと報告されています。しかし、曲げただけで特性が向上する理由は十分に理解されていません。
3次元グラフェンを触媒利用する場合は、「化学反応場となる構造の原子レベルでの精密制御」と「3次元のネットワーク構造全体の安定性」の理解が重要です。化学反応場は、計算資源の都合上、300~1000原子集団のモデルのシミュレーションが一般的であり、300~1000原子集団のモデルから材料全体の特性を精確に理解するのは困難です。また、構造全体の安定性は、局所的な構造から類推するしかない状況です。このような背景から、効率的な触媒設計をするには、上記の制限を理解しかつ経験や勘を総動員し、膨大な選択肢から最適な解を試行錯誤しながら素早く見つけることが求められています。
個人の能力に依存せず普遍的にこれらを達成するために、幾何学にあらわれる「曲率」を考察するとよいことが従来の研究でわかっていました。その中でも「ガウス曲率」(*3)は、構造の内的な歪みをあらわし、化学反応場の幾何学構造の数値化ができます。これらを取り入れた「標準実現」(*4)と呼ばれる数学モデルは直接的また直感的に幾何学構造と材料特性の関係性の理解を提供することが可能で、既存のシミュレーションよりも計算コストがかからないことから、特性の優れたカーボンネットワークの材料探索を加速させると期待されています。
今回、小谷元子教授らの研究グループでは、普遍的かつ貴金属を使用しない触媒材料設計を達成するために、先ほどの標準実現に対して窒素原子などの化学元素を導入することを考えました。より正確に構造を再現するために、近接原子間の反発力を考慮することで、必要最小限の物理現象を組み入れた改善型標準実現モデルを開発し、既存のシミュレーション法を用いて材料設計可能なレベルで構造を再現できていることを確認しました。また、数学モデルから導き出された最適な材料設計を用いて、実際に3次元グラフェンを作製し、意図した触媒設計が有用であることを実験的に実証しました。
研究の内容
3次元グラフェン触媒の設計を行う上で、曲面を形成するために導入された欠陥構造を持つカーボンネットワークの単純化を行いました(図1A)。カーボンネットワークをバネの連結体とみなした標準実現に対して、第二近接原子間の反発力を加えた改善型標準実現を構築しました。この改善型標準実現が正しく構造を構築できるかどうかを最も精度の信頼性が高いとされる第一原理計算手法(*5)と比較して調べました。六員環から構成されるグラフェンの格子に五員環と七員環のペア欠陥(以下、5-7欠陥)を二つ導入した構造を再現したところ、図1Bのように両者の構造特徴は定性的にほぼ一致し、図1Cのように曲面の指標であるガウス曲率の変化を再現できることが明らかになりました。CPU1コアでの計算時間を簡単に比較すると、改善型標準実現の計算時間が39マイクロ秒に対して、第一原理計算手法の計算時間は4万6千秒であったので、改善型標準実現は第一原理計算より10億倍速く計算できることが明らかとなりました。
次に、グラフェンに触媒能力を付与するため、先ほどの5-7欠陥を持つグラフェンモデルの炭素元素1個を窒素元素1個と入れ替えました(ドープしました)。様々な場所の炭素と窒素を入れ替え、これによって変形した構造を詳細に調べました。まず、図2Aと図2Cに示したサイト14の炭素を窒素と入れ替えたところ、改善型標準実現手法と第一原理計算手法で構築された構造の特徴がほぼ一致していることがわかりました。特に、入れ替え前は正のガウス曲率を持っていた箇所が入れ替え後にはほぼゼロの値になっていることが明らかになりました。次に、図2Bと図2Dに示したサイト8の隣の炭素を窒素と入れ替えたところ、改善型標準実現手法と第一原理計算手法で得られた構造の特徴がほぼ一致していることを確認しました。しかし、サイト14のケースとは異なって、入れ替え前後のガウス曲率はほぼ変化していないことが明らかになりました。つまり、入れ替え位置によってガウス曲率が変化する場所と変化しない場所があることがわかりました。このように様々な位置に対して窒素との入れ替えを行い、入れ替え前後のガウス曲率の変化を調査したところ、五員環側では大きな変化があり、六員環が多いところと七員環側では変化がないことが明らかになりました。
ここで、数学的特徴であるガウス曲率に物理的な意味を持たせるため、第一原理計算を用いて各箇所での窒素との入れ替えやすさをエネルギーという物理量で定量的に見積もりました。その結果、窒素との入れ替え前後でガウス曲率の変化量が大きい箇所は窒素と入れ替えるためのエネルギーが小さく、ガウス曲率の変化量が小さい箇所は窒素と入れ替えるためのエネルギーが大きいという相関が得られました。つまり、材料の触媒能力を決める窒素との入れ替えやすさ(ドープしやすさ)は、局所構造を記述するガウス曲率で数値化可能であることがわかりました。これらをまとめると、曲面を形成するためには構造欠陥が存在しなければならず、その欠陥の五員環側の幾何学的歪み(高いガウス曲率)を解消するために異元素が導入されて構造の安定化(ガウス曲率がゼロへ近づく)を図っていると考えられます。
この数学的知見を活かすために触媒活性が高くなるとされる窒素元素と硫黄元素をドープ(入れ替え)した3次元グラフェンを作製しました。図3Aは曲面を持つグラフェンのナノ構造を捉えた電子顕微鏡像で、図3Bはその曲面を拡大した原子構造です。曲面部では、5-7欠陥が観測され、その周囲の構造は六員環構造と窒素や硫黄をドープしたことに起因する欠陥構造が確認されました。次に、異元素がどこに多くドープされているかを調べるために、その場元素マッピングを行いました。図3Cに示したように、矢印がさす平面部は炭素が多く存在しているが窒素や硫黄はほぼ存在せず、丸で囲まれた曲面部は炭素、窒素と硫黄が同時に多く存在していることが明らかになりました。つまり、5-7欠陥周辺に異元素が多く導入されているといえます。
実際に幾何学形状に依存した触媒活性を最先端の走査型電気化学セル顕微鏡(*6)により調べました。0.5 M硫酸水溶液を詰めたナノ電気化学セルプローブを図4Aのように試料表面をなぞるように走査させ、幾何学形状と触媒性能(今回は水素発生能力)を同時に取得しました。図4Bに示したように幾何学形状の起伏が激しい場所で、図4Cに示したような高い還元電流値(該当する触媒反応:2H+ + 2e−à H2)(*7)が観測されました。さらに詳しく見てみると、図4Dや4Eのように幾何学形状の高さが急激に高くなっているところで還元電流値が増大し、平坦部分で逆に還元電流値が小さくなっていることが明らかになりました。つまり、欠陥構造を多く有している曲面部は平面部より水素発生触媒能力が高くなることが実験的に実証されました。
これらの研究成果から、局所構造と構造全体の物性の相関の可視化を実現し、最先端計測技術を用いることで、標準実現手法によって得られた知見が触媒設計に有効であることを実験的に実証することができました。
今後の展開
今回、離散幾何学におけるガウス曲率という特徴がカーボンネットワークの触媒設計に有効であることがわかりましたが、数学的パラメーターと物理的また化学的パラメーターの繋がりの探索は始まったばかりです。今後、網羅的に数学的パラメーターと物理的化学的パラメーターとの関連付けができるように研究を継続する予定です。また、本研究から得られた改善型標準実現手法は幾何学的な手法であることから原理的に無限大のサイズのモデルを計算することができ、既存のシミュレーションと相補的に組み合わせることで原子が1万個を超えるカーボンネットワークに対して大規模計算が可能になります。これらをうまく使い、従来のシミュレーション単独では不可能であった局所構造と大域的な性質をより直接的に結び付けでより正確な材料設計を加速させることが可能な提言ができるよう数学材料連携研究を継続していく予定です。
論文情報
付記事項
本研究成果は、東北大学材料科学高等研究所:Andreas Dechant助教(当時)、大阪大学大学院基礎工学研究科:大戸達彦助教、筑波大学数理物質系:伊藤良一准教授、金沢大学ナノ生命科学研究所:Marina Makarova特任助教、大学院自然科学研究科電子情報科学専攻:河邉佑典博士課程前期学生、理工学域電子情報通信学類:上利龍史学部学生、熊井光学部学生、ナノ生命科学研究所(さきがけ研究者):髙橋康史教授、名古屋大学多元数理科学研究科:内藤久資准教授との共同研究によるものです。
本研究は、JSPS科研費 新学術領域研究「次世代物質探索のための離散幾何学」JP17H06460、 JP17H06465、JP17H06466、 JP18H04477、JP20H04639、JP20H04628、科学研究費助成事業(科研費)、JSTさきがけ(JPMJPR18T8)、世界トップレベル研究拠点プログラム(WPI)、東北大学金属材料研究所における共同研究(新素材共同研究開発センター:202011-CRKEQ-0001、文部科学省委託事業ナノテクノロジープラットフォーム課題として物質・材料研究機構微細構造解析プラットフォーム(課題番号JPMXP09A20NM0013)などの支援を受けて実施されました。
説明図
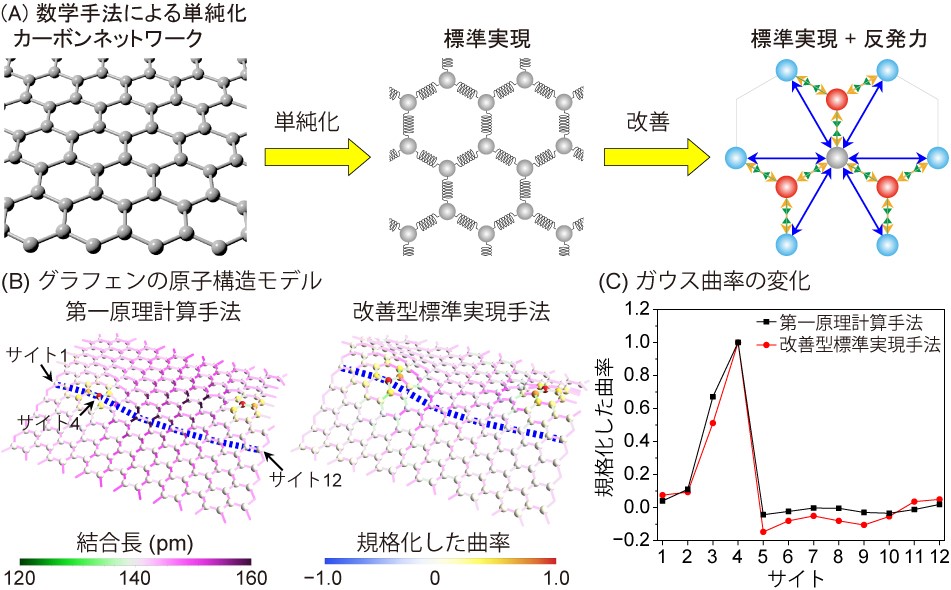
図1. 数学手法によるカーボンネットワークの単純化とグラフェンの原子構造モデル
(A) カーボンネットワークの単純化による標準実現と第2近接同士の反発力を加えた改善型標準実現。
(B) 第一原理計算手法と改善型標準実現手法によって構築されたグラフェンの原子構造モデル。
(C) (B)の青い線に沿って各原子サイトのガウス曲率の変化を取り出した図。
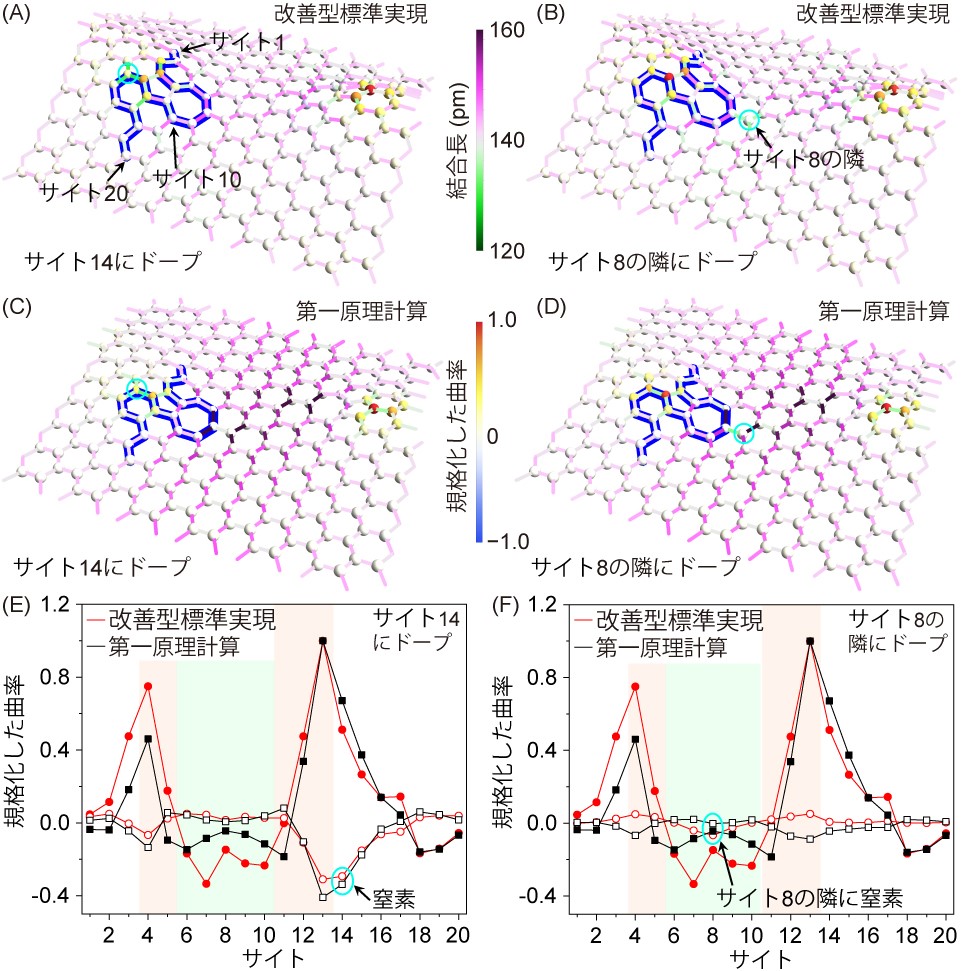
図2. 窒素元素をドープしたグラフェンの原子構造モデルとガウス曲率の変化
(A-B) 完全型標準実現手法を用いたサイト14とサイト8の隣に窒素元素をドープしたグラフェンモデル。
(C-D) 第一原理計算手法を用いたサイト14とサイト8の隣に窒素元素をドープしたグラフェンモデル。
(E-F) サイト14に窒素元素をドープした時のガウス曲率の変化とサイト8の隣に窒素元素をドープした時のガウス曲率の変化。●は窒素ドープ前の規格化したガウス曲率、〇は窒素ドープ後の規格化した曲率から窒素ドープ前の規格化したガウス曲率を差し引いたもの。

図3.曲面を持つグラフェンの原子像と元素マッピング
(A) 低倍率の3次元構造を持つグラフェン像。
(B) 曲面部のグラフェンの高分解像。赤点線は六角形、黄色点線は六角形以外を示す。
(C) その場元素マッピング、炭素C、窒素N、硫黄S、矢印は平面部、丸は曲面部を指す。
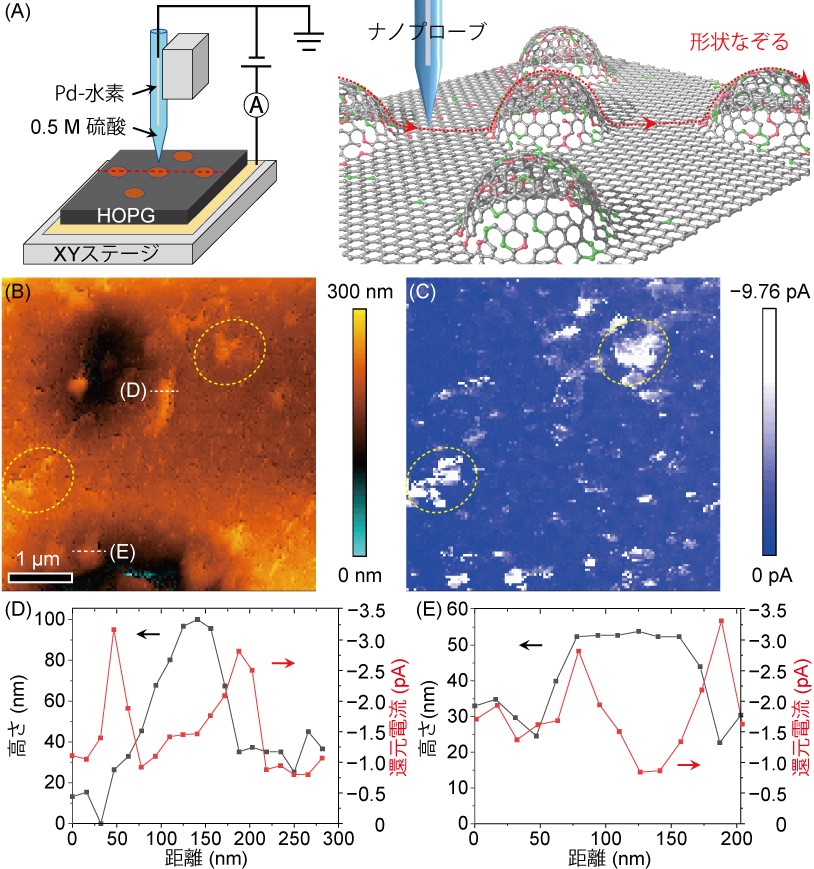
図4. 走査型電気化学セル顕微鏡と還元電流の電気化学イメージング
(A) 走査型電気化学セル顕微鏡の概要図と走査イメージ。
(B) 走査して取得したトポグラフィー図。黄色部は起伏が激しい部分を示している。
(C) 走査して取得した(B)に対する還元電流の電気化学イメージング。
(D-E) (B)における点線の還元電流のラインプロファイル。起伏が激しい部分で還元電流値が大きくなっている。
用語解説
ガウス曲率とは、曲面上の2点間の距離から決まる曲率。ガウス曲率が正である点では、その近くで曲面が接している平面の片側のみに曲面が存在し、負の点では接している平面の両側に曲面が存在する。また、ガウス曲率が0でない曲面上の地図は、2点間の距離を保ったままで平面上の地図に表すことができない。
掲載論文情報
- 論文タイトル
- “Geometric model of 3D curved graphene with chemical dopants” (化学元素をドープした3次元の曲面を持つグラフェンの幾何学モデル)
- 著者
- Andreas Dechant, Tatsuhiko Ohto, Yoshikazu Ito, Marina V. Makarova, Yusuke Kawabe, Tatsufumi Agari, Hikaru Kumai, Yasufumi Takahashi, Hisashi Naito, Motoko Kotani
- 掲載誌
- Carbon
- 掲載日
- 2021.06.04
- DOI
- 10.1016/j.carbon.2021.06.004
- URL
- https://doi.org/10.1016/j.carbon.2021.06.004